What to expect for 2019 with MDR and IVDR implementation?
 Remember the CAMD Roadmap (dating back to end 2017) that promised us a roll-out of MDR and IVDR items that were sometimes even marked ‘high priority’ and how that lifted our spirits (at the time)? Remember how this was supplemented with the Rolling Plan, which promised the roll-out of all roll-outs for 2019?
Remember the CAMD Roadmap (dating back to end 2017) that promised us a roll-out of MDR and IVDR items that were sometimes even marked ‘high priority’ and how that lifted our spirits (at the time)? Remember how this was supplemented with the Rolling Plan, which promised the roll-out of all roll-outs for 2019?
I have great news for you! All action items under the Road Map and the Rolling Plan were just magically delivered!
Oh no, scratch that. Silly joke and way too easy. The Rolling Plan was updated but no delivery of additional items yet. Sorry…
Yet, a lot is happening and a lot is scheduled or bound to happen in 2019 – fasten your seatbelts: here we go.
Updated rolling plan
The updated Rolling Plan was published on 19 February and provides updated timing and next steps for the development of implementing regulations and other actions/initiatives (see here for a nice overview by Medtech Europe):
- MDR Annex XVI products without an intended medical purpose: Consultation on draft text of common specifications in Q1 2019 now; dates for finalisation has moved up to Q1 2020.
- ‘Scientific bodies’ (i.e., expert panels, EU reference laboratories and expert laboratories): For the various implementing acts the surveys are now marked as ‘finalised’ (as in December 2018). Only change: the move into drafting stage for the act on expert panels.
- Eudamed: still under construction – EUDAMED for PMS and clinicals may be only partly available or not at all by the date of application.
- Communications campaign: Documents slowly trickle in on the new dedicated website and library; public awareness initiatives underway.
- Medical Device Coordination Group (MDCG) subgroups: the evaluation of the applications is still ongoing, new MDCG subgroups are expected in Q1 2019.
- EU Medical devices nomenclature – the Commission is currently finalising its assessment in the view of a final decision to be taken by Q1 2019.
- Notified Body designation: 42 applications received by the EC, 3 further joint assessments scheduled (apart from the 25 already carried out).
Everything else stays the same. Summary: everything not delivered so far will be delivered in 2019 and if it isn’t delivered in 2019 there is a problem. Except for Eudamed, which is scheduled for March 2020 still, but in less functionality than originally expected. You’ll be able to enter your your devices though – hopefully.
Don’t mention the Brexit
Except that we must – the EU27 national authorities are now giving their own individual interpretations of what a Brexit scenario entails for labelling and placing on the market of products and it does not look pretty.
The Dutch authority has for example said that NBOG 2006-1 is out of the window and companies must amend their labelling per Brexit date, if necessary by means of stickering (which, as the drama in UK Parliament continues and everybody has lost patience with all the political posturing and delaying, may well be end of this month). This uncertainty is not a happy place to be in for companies selling medical devices.
Yes dear readers, the word you are looking for starts with ‘cluster’ and rhymes with ‘duck’. It shows us what a special breed of extraordinarily responsible persons politicians can be. We will miss the MHRA though, and all the good work they did in driving many of the MDR and IVDR work items at EU level – I personally am sad to see them go.
Upcoming corrigendum
 There is the interesting matter of the MDR and IVDR corrigendum coming our way. This publication is supposed to fix the mistakes and inconsistencies in these regulations and is a normal instrument for the EU that is regularly used for regulation that turns out to have mistakes in it. The big speculation of course is whether other things are changed too. While this is constitutionally not possible (as we would need to follow the legislative procedure for amending legislation for this) it may be that nevertheless some things will be sneaked into the amended texts.
There is the interesting matter of the MDR and IVDR corrigendum coming our way. This publication is supposed to fix the mistakes and inconsistencies in these regulations and is a normal instrument for the EU that is regularly used for regulation that turns out to have mistakes in it. The big speculation of course is whether other things are changed too. While this is constitutionally not possible (as we would need to follow the legislative procedure for amending legislation for this) it may be that nevertheless some things will be sneaked into the amended texts.
I have seen a glimpse from what the corrigendum will contain in terms of non-corrections. The French competent authority ANSM published a report from a meeting with industry in which it went on the record that:
“The Commission has prepared a corrigendum to the regulations on MDR and IVDR to be presented to the Council and Parliament. In addition to simple editorial corrections, it is proposed to extend the transitional measures to Class I devices that require the use of a notified body (NB) or Class I devices that change class under the MDR, in order to avoid overloading NBs. While a large majority of the competent authorities are in favour of this measure, there are differences as to its legal form.
SIDIV asked whether such a measure was being considered for IVDs. For the time being, this is not the case, as the IVDR has a 5-year application period from its entry into force.” (my own translation from French)
If things happen this way (big if) then this would be big break for the struggling class I devices companies (like for example software) that I see have great difficulties finding notified body capacity for the MDR (because – duh, not there yet). I am personally not sure what the corrigendum will look like in the end, because the process is rather secretive and will likely be subject to some last minute politically motivated course corrections.
The ANSM report also confirms the difficulties that many companies are having with their notified bodies (which I have flagged repeatedly) and states that this is a grave concerned of ANSM. ANSM also confirms the widespread nature of notified bodies failing to meet their obligations throughout Europe, invites industry to notify it of any such practices and states that the Commission has been advised of this.
Upcoming guidance and implementation
Expect more guidance being delivered this summer. By mid-2019:
- Guidance on vigilance (maybe earlier than summer)
- Guidance on software
- Guidance on classification
- Guidance on clinical evidence
Further guidance is under preparation:
- Guidance for substance-based devices
- Common Specifications on Annex XVI (consultation ongoing at the moment)
- Common Specifications on reprocessing (as well as Implementing Act)
Delivery of all of these items would allow for some good progress to be made under the CAMD Roadmap, so fingers crossed!
EMA guidance on combination products
The EMA has recently released a new guidance note on combination products in relation to the MDR and IVDR. As the EMA explains, important stuff because one in four centrally authorised medicines contains a device component. This first guidance note focuses on article 117 Directive 2001/83 because (yes it’s true) the MDR and IVDR amended the Medicinal Products Directive and applications for a marketing authorisation of a medicinal product with an integral medical device submitted as of 26 May 2020 must comply with the requirements of Article 117 of Regulation 2017/745. Article 117 MDR does not affect existing MAs for combination products but if there is a substantial change to the design or intended purpose of the device component, or a new device is introduced, after authorisation any required certificate/declaration of conformity/NB opinion should be submitted (as appropriate) to EMA/NCA as part of the variation/extension application.
The guidance will be continuously updated with new questions and new answers.
Eudamed specs
The Commission published a first draft version of the Eudamed specifications. One of the interesting things for me was that it became clear to what extent the database is open to the public. Section 8.2 of the document sets out the database fields that are available to the public: economic actor details UDI/device details, certificate and notified body details, information concerning clinical investigations (including reports and summaries), vigilance data and PMS information and market surveillance details. Confidential information will not be made public in these fields but to me it is as yet completely unclear how that will be arranged – the document refers to use of summaries and hiding of confidential data but that is not very precise. With respect to vigilance data the document states:
“To determine [what data remains confidential], it is to be defined what vigilance and post-market surveillance information could be available to the public. As long as there is no agreement among MS and with the Commission, these data will not be publically available.”
It will be interesting to see on what level of publicity this discussion will land. I hope that the Commission and authorities realise that there is legislation for personal data (GDPR) that is relevant to situations where there are small groups of patients concerned, and that too far going publication may negatively manufacturer willingness to report, as vigilance reports may often contain confidential details about devices.
More Eudamed and UDI related news: the designation of UDI issuing entities is expected by May 2019.
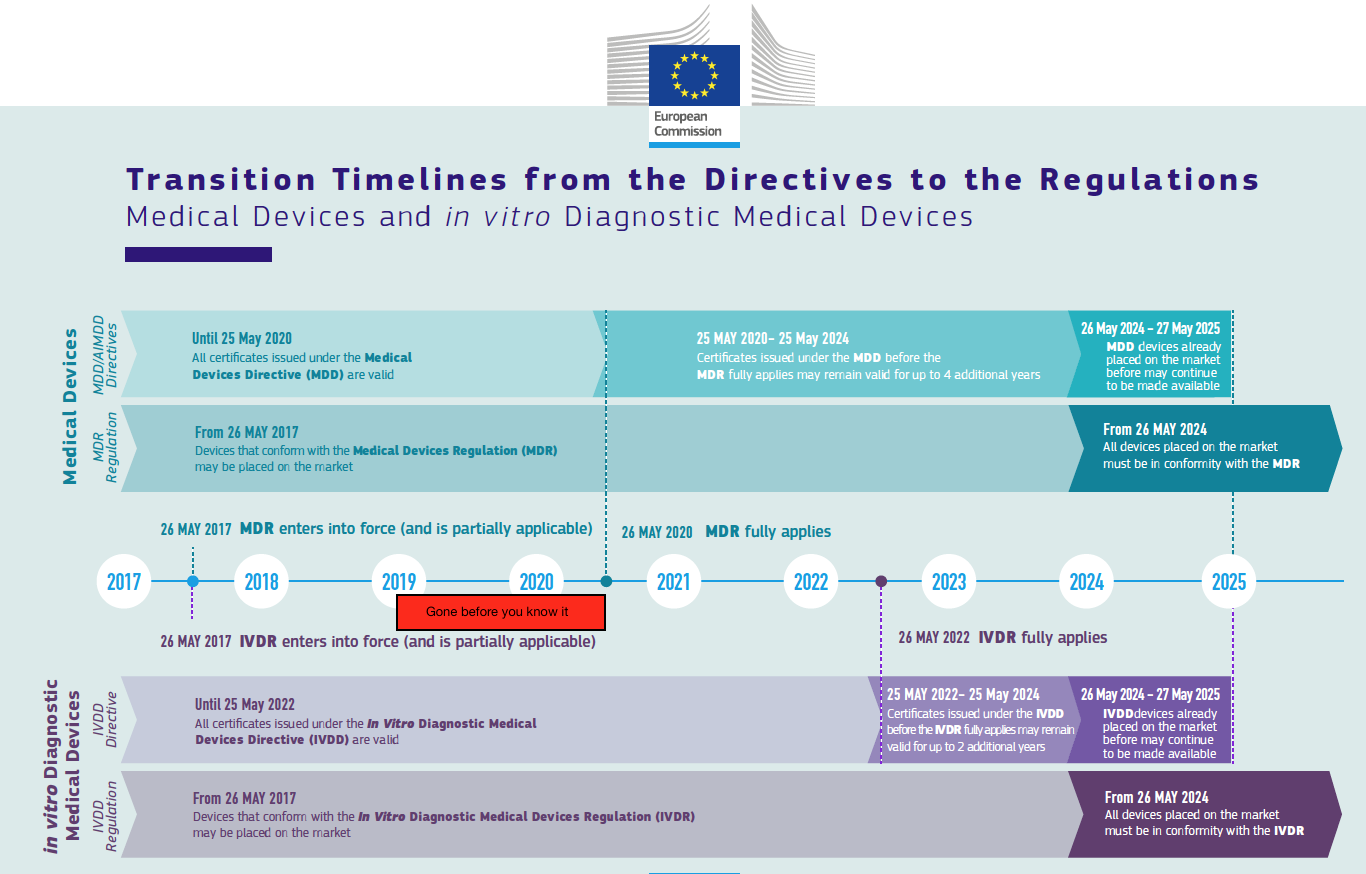
2019, the year of soft transition deadlines
Finally, 2019 will be the year of re-certification under the old directives one more year to benefit from soft transition. If you are planning to use this option, notified bodies are going to ask you to submit an application somewhere this year. Some are very clear about their deadlines (BSI has for example said end of Q1 2019), while others may be somewhat more flexible.
So 2019 will be the year of choices: are you going for recertification or the new regulation certificate, and in the first case: how will you not miss your window? The closer to May 2020, the busier the notified bodies will be with the MDR certifications, and they want the (AI)MDD re-certifications done before that time. Do not count on being able to do (AI)MDD recertification as in 2020, unless your notified body is very very confident that they have the capacity to do this.
Finally a podcast
Feeling a bit overwhelmed by all this detail? In that case zoom out to the bigger picture and hear about the background to these new regulations on the Medical Devices Made Easy podcast.